

مشخصات تست بررسی جهشهای شایع ژن GBA در بیماری گوشه
بیماریهای مرتبط:
ناهنجاریهای پوستی، بزرگی کبد و طحال، بیماریهای سیستم عصبی، کمخونی، ترومبوسیتوپنی، بیماریهای ریوی، هپاتومِگالی
روش انجام آزمایش:
Reverse Strip Assay
نوع نمونه:
5 - 10 سیسی خون محیطی با ضد انعقاد EDTA
2 سرنگ هرکدام به میزان 10 سیسی نمونه مایع آمنیوتیک یا حداقل 10 میلیگرم نمونه پرز جفتی (برای تشخیص پیش از تولد* با هماهنگی قبلی)
(*تشخيص پیش از تولد صرفا در شرایطی قابل انجام است که وضعيت زوجين از نظر ناقل بودن مشخص شده باشد)
زمان پاسخدهی:
14 روز کاری
(در صورت ارسال نمونه مایع آمنیوتیک برای تشخیص پیش از تولد، 10 الی 14 روز به زمان جوابدهی آزمایش افزوده خواهد شد)
مدارک مورد نیاز:
تکمیل فرم ثبت اطلاعات آزمایشات تشخیص پس از تولد
رسم شجرهنامه یا ارائه سابقه بیماری در خانواده، ارائه اطلاعات بالينی و مدارک پزشکی مرتبط با بيماری
درصورت درخواست آزمایش برای تشخیص پیش از تولد، تکمیل فرم ثبت اطلاعات آزمایشات تشخیص پیش از تولد
دستورالعملها:
دانلود پروتکل نحوه تهیه و ارسال نمونه خون محیطی با ضد انعقاد EDTA
دانلود معیارهای پذیرش و نحوه برخورد با موارد مشکلدار نمونه خون محیطی
خدمات مرتبط
[
{
"serviceId": 10,
"namePersian": "آزمایش ژنتیک برای تشخیص بیماری SMA",
"nameArabic": "الفحص الجيني لتشخيص مرض SMA",
"nameEnglish": "Genetic Testing for SMA Diagnosis",
"slug": null,
"serviceType": "laboratory",
"iconFileId": null,
"iconFile": null,
"coverFileId": 263,
"coverFile": null,
"descriptionPersian": "آتروفی عضلانی نخاعی، یک بیماری ارثی عصبی عضلانی",
"descriptionArabic": " ضمور العضلات الشوكي، مرض عصبي عضلي وراثي",
"descriptionEnglish": " Spinal Muscular Atrophy, a Hereditary Neuromuscular Disease",
"overviewPersian": "<p style=\"text-align: justify; line-height: 2;\">آتروفی عضلانی نخاعی (SMA) بیماری ارثی است که باعث تحلیل و ضعف پیشرونده عضلات درگیر در بسیاری از عملکردهای اساسی مانند تنفس، حرکت، خوردن و ... می‌شود. افراد مبتلا به SMA نوع خاصی از سلول‌های عصبی نخاع به نام نورون‌های حرکتی را از دست می‌دهند که حرکت عضلات را کنترل می‌کنند. بدون این نورون‌های حرکتی، عضلات سیگنال‌های عصبی را که باعث حرکت آنها می‌شود را دریافت نمی‌کنند. این بیماری معمولاً با الگوی اتوزومی مغلوب به فرزندان منتقل می‌شود، بدین معنی که فرد باید ژن معیوب را از هم از پدر و هم از مادر دریافت کند تا تحت تأثیر این بیماری قرار گیرد. </p>\n<p><img style=\"display: block; margin-left: auto; margin-right: auto;\" src=\"https://webapi.irangenepath.com/files/view/2024/04/06/265/14af128d783f4da29d152c365726d0d1.png\" alt=\"\" width=\"300\" height=\"427\" /></p>\n<p style=\"text-align: justify; line-height: 2;\">انواع مختلفی از SMA وجود دارد که تیپ یا زیرگروه نامیده می‌شوند و بر اساس شدت اختلال و سن شروع علائم تعیین می‌گردند. به طور کلی، سه نوع SMA وجود دارد که کودکان را قبل از 1 سالگی تحت تأثیر قرار می‌دهد و دو نوع SMA نیز وجود دارد که در بزرگسالی و معمولاً پس از 30 سالگی اتفاق می‌افتد. اغلب انواع این بیماری به دلیل بروز جهش‌ در ژن SMN1 ایجاد می‌شوند که منجر به سطح ناکافی بیان پروتئین SMN می‌گردد. برای تشخیص آتروفی عضلانی نخاعی، در کنار بررسی علائم این بیماری، از طریق انجام آزمایش ژنتیک می‌توان بیماری را تشخیص داد. آزمایش بررسی حذف‌های ژنی در ژن SMN1 و همچنین SMN2 در بیماران مشکوک به آتروفی عضلانی نخاعی و همچنین بررسی جنین در هنگام بارداری با هدف تشخیص پیش از تولد آتروفی عضلانی نخاعی مورد استفاده قرار می‌گیرد.</p>",
"overviewTypePersian": "html",
"overviewArabic": "",
"overviewTypeArabic": null,
"overviewEnglish": "",
"overviewTypeEnglish": null,
"inpersonPrice": null,
"telPrice": null,
"onlineTextPrice": null,
"onlineVoicePrice": null,
"onlineVideoPrice": null,
"onsitePrice": null,
"reviewCount": 0,
"reviewRate": null,
"views": 0,
"isActive": true,
"tags": [
{
"tagId": 24,
"name": "بیماری ژنتیکی",
"uses": 0,
"createdAt": "2024-03-10T09:29:40.000Z",
"updatedAt": "2024-03-10T09:29:40.000Z"
}
],
"createdAt": "2024-01-01T11:26:12.000Z",
"updatedAt": "2024-04-06T10:11:42.000Z"
},
{
"serviceId": 8,
"namePersian": "بررسی وضعیت ناقلیت در خانم و آقا",
"nameArabic": "فحص حالة الناقل للرجال والنساء",
"nameEnglish": "Carrier Status Testing for Men and Women",
"slug": null,
"serviceType": "laboratory",
"iconFileId": null,
"iconFile": null,
"coverFileId": 266,
"coverFile": null,
"descriptionPersian": "پنل اختصاصی تعیین وضعیت ناقلیت فرد در 433 ژن همراه با بررسی تغییرات ژنتیکی در بیماریهای Fragile X ، SMA و Duchenne",
"descriptionArabic": " لوحة خاصة لتحديد حالة الناقل في 433 جين مع تحليل التغييرات الجينية في أمراض Fragile X و SMA و Duchenne",
"descriptionEnglish": " Special Panel for Carrier Status Determination in 433 Genes with Genetic Changes Analysis in Fragile X, SMA, and Duchenne Diseases",
"overviewPersian": "<p style=\"line-height: 2; text-align: justify;\">همه افراد، حتی در صورت منفی بودن سابقه خانوادگی اختلال ژنتیکی، می‌توانند حامل جهش‌های ژنتیکی باشند که پتانسیل ایجاد اختلالات ژنتیکی را داشته باشد. حامل یک جهش ژنتیکی بودن همیشه به معنای ابتلای فرد به بیماری ژنتیکی نیست، بلکه غالبا به این معنی است که ممکن است با انتقال این جهش‌های ژنتیکی از والدین، فرزندان آن‌ها در معرض خطر ابتلا به یک اختلال ژنتیکی قرار گیرند. بررسی وضعیت ناقلی یک آزمایش ژنتیکی است که بواسطه آن می‌توان تشخیص داد آیا یک فرد سالم، ناقل یک بیماری ژنتیکی با توارث اتوزم مغلوب و یا وابسته به جنس است یا خیر، و هدف از انجام این آزمایش، کمک به زوج‌ها برای آرامش خاطر در دوران بارداری و کاهش ریسک تولد فرزند مبتلا به یک اختلال ژنتیکی است.</p>\n<p style=\"line-height: 2; text-align: justify;\">از آنجاییکه نرخ ازدواج‌های خویشاوندی در کشور ما نسبتا بالا است، ریسک ناقلیت این گروه از بیماری‌ها نیز بیشتر است. برای مثال مطالعات انجام شده در کشورمان در سال‌های گذشته نشان داده است که از هر 20 نفر، حداقل یک نفر ناقل بیماری SMA می‌باشد. همچنین مطالعه‌ای بر روی حدود 23500 نمونه از قومیت‌های مختلف از نظر بررسی 417 جهش بیماری‌زا که سبب 108 بیماری اتوزم مغلوب می‌شوند انجام شد. نتایج این مطالعه نشان داد که از هر 100 نفر 24 نفر ناقل حداقل یکی از 108 بیماری مورد بررسی بودند. همچنین مطالعه آنها نشان داد که از هر 100 نفر تقریبا 5 نفر ناقل چند بیماری بوده‌اند.</p>\n<p style=\"line-height: 2;\"><img style=\"display: block; margin-left: auto; margin-right: auto;\" src=\"https://webapi.irangenepath.com/files/view/2024/04/06/267/8905fa517de04e0f92ddb8688342cd3a.png\" alt=\"\" width=\"800\" height=\"256\" /></p>\n<p style=\"line-height: 2; text-align: justify;\">انجام این آزمایش به همه زوج‌هایی که قصد بارداری دارند توصیه می‌گردد، همچنین زوج‌هایی با شرایط ذیل، به عنوان کاندید قطعی برای انجام این آزمایش در نظر گرفته می‌شوند:</p>\n<ul style=\"text-align: justify;\">\n<li style=\"line-height: 2;\">زوج‌هایی با سابقه خانوادگی یا دارای فرزند مبتلا به بیماری ژنتیکی</li>\n<li style=\"line-height: 2;\">زوج‌هایی با نسبت خویشاوندی (ازدواج فامیلی)</li>\n<li style=\"line-height: 2;\">زوج‌هایی از قومیت‌هایی با شیوع بالای بیماری‌های ژنتیکی خاص</li>\n<li style=\"line-height: 2;\">زوج‌هایی که قصد بارداری با روش‌های کمک باروری مانند IVF داشته باشند.</li>\n</ul>\n<p style=\"line-height: 2; text-align: justify;\">برای دستیابی به نتایج بهتر و کاربردی از این آزمایش، توصیه می‌شود که زوجین، هر دو، نسبت به انجام این تست اقدام نمایند. این تست به منظور تعیین وضعیت ناقل بودن فرد برای تعداد 433 ژن موجود در پانل که مسبب یک یا چند اختلال ژنتیکی با توارث اتوزم مغلوب (autosomal recessive) و وابسته به جنس (X-linked) هستند، براساس گایدلاین‌های انجمن ژنتیک‌پزشکی و ژنومیک آمریکا (ACMG) طراحی شده است.</p>",
"overviewTypePersian": "html",
"overviewArabic": "",
"overviewTypeArabic": null,
"overviewEnglish": "",
"overviewTypeEnglish": null,
"inpersonPrice": null,
"telPrice": null,
"onlineTextPrice": null,
"onlineVoicePrice": null,
"onlineVideoPrice": null,
"onsitePrice": null,
"reviewCount": 0,
"reviewRate": null,
"views": 0,
"isActive": true,
"tags": [
{
"tagId": 24,
"name": "بیماری ژنتیکی",
"uses": 0,
"createdAt": "2024-03-10T09:29:40.000Z",
"updatedAt": "2024-03-10T09:29:40.000Z"
}
],
"createdAt": "2024-01-01T09:57:48.000Z",
"updatedAt": "2024-04-06T10:48:43.000Z"
},
{
"serviceId": 7,
"namePersian": "بررسی ژنتیکی بیماری سلیاک",
"nameArabic": "الفحص الجيني لمرض السيلياك",
"nameEnglish": "Genetic Testing for Celiac Disease",
"slug": null,
"serviceType": "laboratory",
"iconFileId": null,
"iconFile": null,
"coverFileId": 268,
"coverFile": null,
"descriptionPersian": "بررسی ژنهای موثر در ایجاد بیماری سلیاک",
"descriptionArabic": " تحليل الجينات المساهمة في مرض السيلياك",
"descriptionEnglish": " Analysis of Genes Contributing to Celiac Disease",
"overviewPersian": "<p style=\"text-align: justify; line-height: 2;\">بیماری سلیاک یک اختلال گوارشی است و به عنوان واکنش ایمنی بدن در برابر خوردن گلوتن بیان می‌شود. گلوتن پروتئینی است که در گندم، جو و چاودار (گندم سیاه) و تمامی محصولات حاوی این نوع غلات یافت می‌شود. هنگامی که فرد مبتلا به سلیاک محصولی حاوی گلوتن می‌خورد، بدن او بیش از حد به این پروتئین واکنش نشان داده و با گذشت زمان به پوشش روده کوچک آسیب رسانده و از جذب برخی مواد مغذی توسط آن جلوگیری می‌کند. در نهایت، می‌تواند منجر به سوء تغذیه و همچنین از دست دادن تراکم استخوان، سقط جنین، ناباروری یا حتی بیماری‌های عصبی یا برخی سرطان‌ها شود. اختلال در ژن‌ها در کنار خوردن غذاهای حاوی گلوتن و برخی عوامل دیگر می‌توانند در ایجاد بیماری سلیاک نقش داشته باشند، اما علت دقیق آن هنوز مشخص نیست. عفونت‌های دستگاه گوارش و باکتری‌های روده نیز ممکن است در این امر نقش داشته باشد. گاهی بیماری سلیاک پس از جراحی، بارداری، زایمان، عفونت ویروسی یا فشار روحی شدید نیز فعال می‌شود.</p>\n<p><img style=\"display: block; margin-left: auto; margin-right: auto;\" src=\"https://webapi.irangenepath.com/files/view/2024/04/06/269/e4aa1761544d4fcaae4f4594c04c5306.jpeg\" alt=\"\" width=\"500\" height=\"253\" /></p>\n<p style=\"text-align: justify; line-height: 2;\">علائم و نشانه‌های بیماری سلیاک می‌تواند در کودکان و بزرگسالان بسیار متفاوت باشد. علائم و نشانه‌های گوارشی برای بزرگسالان عبارتند از: اسهال، خستگی، درد شکم، کاهش وزن، نفخ شکم، تهوع و استفراغ و یبوست. با این حال، بیش از نیمی از بزرگسالان مبتلا به بیماری سلیاک علائم و نشانه‌هایی دارند که با دستگاه گوارش ارتباط ندارند، از جمله: کم خونی، معمولا ناشی از کمبود آهن، پوکی یا نرم شدن استخوان، بثورات پوستی خارش‌دار یا تاول‌دار، زخم‌های دهانی، سردرد و خستگی، آسیب سیستم عصبی و درد مفاصل. در کودکان مبتلا به سلیاک نیز علاوه بر علائمی که در بزرگسالان دیده می‌شود، می‌تواند بر رشد و نمو تأثیر بگذارد و همچنین آنها بیشتر از بزرگسالان دچار مشکلات گوارشی مانند تهوع و استفراغ، اسهال مزمن، تورم شکم، یبوست، نفخ شکم و مدفوع کم رنگ و بد بو می‌شوند.</p>\n<p style=\"text-align: justify; line-height: 2;\">بیماری سلیاک یک اختلال چند ژنی است که در آن غالب‌ترین عوامل خطر ژنتیکی، انواع خاص HLA-DQ هستند. (<span style=\"font-family: tahoma, arial, helvetica, sans-serif;\">DQ2</span> و <span style=\"font-family: tahoma, arial, helvetica, sans-serif;\">DQ8</span>) توسط ناحیه HLA-class II در کروموزوم 6 کدگذاری شده است. از آنجایی که اکثریت افراد حامل آلل‌های مرتبط با بیماری سلیاک هستند، اما تحت تاثیر این بیماری قرار نمی‌گیرند، بنابراین نمی‌توان از نتیجه مثبت این آزمایش به عنوان پارامتر قطعی برای تشخیص ابتلا به بیماری سلیاک استفاده کرد.</p>",
"overviewTypePersian": "html",
"overviewArabic": "",
"overviewTypeArabic": null,
"overviewEnglish": "",
"overviewTypeEnglish": null,
"inpersonPrice": null,
"telPrice": null,
"onlineTextPrice": null,
"onlineVoicePrice": null,
"onlineVideoPrice": null,
"onsitePrice": null,
"reviewCount": 0,
"reviewRate": null,
"views": 0,
"isActive": true,
"tags": [
{
"tagId": 24,
"name": "بیماری ژنتیکی",
"uses": 0,
"createdAt": "2024-03-10T09:29:40.000Z",
"updatedAt": "2024-03-10T09:29:40.000Z"
}
],
"createdAt": "2024-01-01T08:19:50.000Z",
"updatedAt": "2024-04-06T12:29:14.000Z"
},
{
"serviceId": 6,
"namePersian": "تشخیص بیماری تب مدیترانهای",
"nameArabic": "تشخيص حمى البحر المتوسط",
"nameEnglish": "Mediterranean Fever Diagnosis",
"slug": null,
"serviceType": "laboratory",
"iconFileId": null,
"iconFile": null,
"coverFileId": 270,
"coverFile": null,
"descriptionPersian": "بررسی ژنتیکی احتمال ابتلا به بیماری FMF",
"descriptionArabic": " الفحص الجيني لاحتمالية الإصابة بمرض FMF",
"descriptionEnglish": " Genetic Testing for FMF Disease Risk",
"overviewPersian": "<p style=\"text-align: justify; line-height: 2;\">تب مدیترانه‌ای فامیلیال (FMF) یک بیماری ژنتیکی خود التهابی است که باعث تب‌های مکرر و التهاب دردناک شکم، ریه‌ها و مفاصل و بثورات پوستی می‌شود. این بیماری یک اختلال ارثی است که اغلب در افرادی بروز پیدا می‌کند که اجداد آنها در کشورهای اطراف دریای مدیترانه و خاورمیانه زندگی می‌کردند. تب مدیترانه‌ای معمولاً در دوران کودکی تشخیص داده شده و تقریباً همه افراد مبتلا به FMF قبل از 20 سالگی دچار تب‌های دوره‌ای می‌شوند این بیماری به دلیل بروز جهش در ژنی به نام MEFV ایجاد می‌شود و کودکان مبتلا معمولاً نسخه‌ای از ژن معیوب MEFV را از والدین به ارث می‌برند. بسیاری از جهش‌های مختلف در ژن MEFV با بیماری تب مدیترانه‌ای مرتبط هستند. برخی جهش‌ها ممکن است موارد بسیار شدیدی از بیماری ایجاد کنند، در حالی که برخی دیگر ممکن است علائم و نشانه‌های خفیف‌تری داشته باشند.</p>\n<p style=\"line-height: 2;\"><img style=\"display: block; margin-left: auto; margin-right: auto;\" src=\"https://webapi.irangenepath.com/files/view/2024/04/06/271/ef761e02423d48debfd784b32b42ff1f.jfif\" alt=\"\" width=\"500\" height=\"332\" /></p>\n<p style=\"text-align: justify; line-height: 2;\">علائم و نشانه‌های تب مدیترانه‌ای معمولاً در دوران کودکی شروع می‌شود. علائم معمولا یک تا سه روز طول می‌کشد. علاوه بر تب، درد و تورم مفاصل معمولاً در زانو یا مچ پا نیز دیده می‌شود. مفصل ممکن است به اندازه‌ای دردناک و متورم باشد که مانع از راه رفتن کودک شود. بیمار مبتلا به تب مدیترانه‌ای فامیلیال یا FMF ممکن است این علائم را تجربه کند:</p>\n<ul>\n<li style=\"text-align: justify;\">تب‌های عود کننده</li>\n<li style=\"text-align: justify;\">درد و تورم مفاصل</li>\n<li style=\"text-align: justify;\">بثورات قرمز رنگ روی ساق و مچ پا</li>\n<li style=\"text-align: justify;\">دردهای شدید شکمی</li>\n<li style=\"text-align: justify;\">درد قفسه سینه</li>\n<li style=\"text-align: justify;\">درد در ماهیچه‌ها</li>\n</ul>\n<p style=\"text-align: justify; line-height: 2;\">از آنجایی که جهش‌های ژنی ایجاد کننده این بیماری شناسایی شده‌اند، با بررسی ژن MEFV می‌توان این بیماری را تشخیص داد که این آزمایش در آزمایشگاه پاتولوژی و ژنتیک کریمی‌نژاد - نجم‌آبادی قابل انجام است.</p>\n<p style=\"text-align: justify; line-height: 2;\"> </p>",
"overviewTypePersian": "html",
"overviewArabic": "",
"overviewTypeArabic": null,
"overviewEnglish": "",
"overviewTypeEnglish": null,
"inpersonPrice": null,
"telPrice": null,
"onlineTextPrice": null,
"onlineVoicePrice": null,
"onlineVideoPrice": null,
"onsitePrice": null,
"reviewCount": 0,
"reviewRate": null,
"views": 0,
"isActive": true,
"tags": [
{
"tagId": 24,
"name": "بیماری ژنتیکی",
"uses": 0,
"createdAt": "2024-03-10T09:29:40.000Z",
"updatedAt": "2024-03-10T09:29:40.000Z"
}
],
"createdAt": "2024-01-01T08:08:52.000Z",
"updatedAt": "2024-04-06T12:58:24.000Z"
},
{
"serviceId": 1,
"namePersian": "کاریوتایپ خون محیطی",
"nameArabic": " النمط النووي للدم المحيطي",
"nameEnglish": " Peripheral Blood Karyotype",
"slug": "",
"serviceType": "laboratory",
"iconFileId": 0,
"iconFile": null,
"coverFileId": 280,
"coverFile": null,
"descriptionPersian": "تشخیص ناهنجاریهای کروموزومی در خانمها و آقایان",
"descriptionArabic": " تشخيص الشذوذات الكروموسومية في الرجال والنساء",
"descriptionEnglish": " Chromosomal Abnormalities Diagnosis in Men and Women",
"overviewPersian": "<p class=\"MsoNormal\" dir=\"RTL\" style=\"text-align: justify; direction: rtl; unicode-bidi: embed; line-height: 2;\"><span lang=\"FA\" style=\"font-family: IRANYekan, sans-serif;\">انسان‌ها به طور طبیعی دارای 46 کروموزم هستند که به صورت 23 جفت کروموزوم وجود دارند. از این 23 جفت کروموزوم، 22 جفت کروموزوم غیرجنسی (اتوزوم) هستند و یک جفت جفت باقیمانده از کروموزوم‌های جنسی تشکیل شده است که جنسیت مرد (</span><span dir=\"LTR\" style=\"font-family: IRANYekan, sans-serif;\">XY</span><span lang=\"FA\" style=\"font-family: IRANYekan, sans-serif;\"> - مرد) یا زن (</span><span dir=\"LTR\" style=\"font-family: IRANYekan, sans-serif;\">XX</span><span lang=\"FA\" style=\"font-family: IRANYekan, sans-serif;\"> - زن) را تعیین می‌کند. </span><span style=\"font-family: IRANYekan, sans-serif;\">به طور معمول، تمام سلول‌های بدن که دارای هسته هستند دارای مجموعه کاملی از همان 46 کروموزوم هستند، به جز سلول‌های تولیدمثلی (تخمک و اسپرم)، که شامل نیم مجموعه از 23 جفت یا به عبارت دیگر 23 کروموزم می‌باشند که در زمان لقاح هر نیم مجموعه از پدر و از مادر به فرزند منتقل و باهم ترکیب می‌شوند و مجموعه جدیدی از 46 کروموزوم یا 23 جفت کروموزوم را در جنین در حال رشد تشکیل می‌دهند.</span></p>\n<p class=\"MsoNormal\" dir=\"RTL\" style=\"text-align: justify; direction: rtl; unicode-bidi: embed; line-height: 2;\"><span style=\"font-family: IRANYekan, sans-serif; font-size: 10pt;\">ناهنجاری‌های کروموزومی شامل تغییرات تعدادی و ساختاری است. برای تغییرات تعدادی، هر چیزی غیر از مجموعه کاملی از 46 کروموزوم نشان دهنده تغییر در مقدار مواد ژنتیکی موجود است و می‌تواند باعث ناهنجاری‌ها و اختلالات رشد شود. در تغییرات ساختاری، اهمیت ناهنجاری‌ها و شدت آنها به کروموزوم تغییر یافته بستگی دارد. در واقع نوع و درجه ناهنجاری می‌تواند از فردی به فرد دیگر متفاوت باشد، حتی اگر ناهنجاری کروموزومی یکسانی وجود داشته باشد.</span></p>\n<p class=\"MsoNormal\" dir=\"RTL\" style=\"direction: rtl; unicode-bidi: embed; line-height: 2;\"><img style=\"display: block; margin-left: auto; margin-right: auto;\" src=\"https://webapi.irangenepath.com/files/view/2024/04/07/281/dc63744c544043cbb940f51a9696b81f.jfif\" alt=\"\" width=\"400\" height=\"293\" /></p>\n<p class=\"MsoNormal\" dir=\"RTL\" style=\"text-align: justify; direction: rtl; unicode-bidi: embed; line-height: 2;\"><span style=\"font-family: IRANYekan, sans-serif;\">مطالعه کروموزومی یا کاریوتایپ آزمایشی است که تعداد و ساختار کروموزوم‌های فرد را به منظور تشخیص ناهنجاری‌های احتمالی ارزیابی می‌کند. </span><span style=\"font-family: IRANYekan, sans-serif;\">این آزمایش معمولاً در افراد دارای مشکلاتی از قبیل ناباروری، سقط مکرر، سابقه خانوادگی اختلالات کروموزمی، تأخیر در رشد، ناهنجاری‌های مادرزادی، ناتوانی‌های ذهنی و جسمی، شرایط جسمانی و ظاهری غیرطبیعی و ... انجام می‌شود. </span><span style=\"font-family: IRANYekan, sans-serif;\">بدیهی است که انجام صحیح آزمایش و تفسیر درست نتایج به تجربه و تخصص نیاز دارد و مرکز پاتولوژی و ژنتیک کریمی‌نژاد - نجم‌آبادی به عنوان اولین مرکز تشخیص پیش از تولد در کشور، با سابقه انجام بیش از 250 هزار کاریوتایپ آماده ارائه خدمات در این زمینه به مراجعین عزیز می‌باشد.</span></p>",
"overviewTypePersian": "html",
"overviewArabic": "",
"overviewTypeArabic": null,
"overviewEnglish": "",
"overviewTypeEnglish": null,
"inpersonPrice": null,
"telPrice": null,
"onlineTextPrice": null,
"onlineVoicePrice": null,
"onlineVideoPrice": null,
"onsitePrice": null,
"reviewCount": 0,
"reviewRate": null,
"views": 0,
"isActive": false,
"tags": [
{
"tagId": 24,
"name": "بیماری ژنتیکی",
"uses": 0,
"createdAt": "2024-03-10T09:29:40.000Z",
"updatedAt": "2024-03-10T09:29:40.000Z"
}
],
"createdAt": "2023-03-29T11:45:53.000Z",
"updatedAt": "2025-12-17T08:19:53.000Z"
}
] پنل اختصاصی تعیین وضعیت ناقلیت فرد در 433 ژن همراه با بررسی تغییرات ژنتیکی در بیماریهای Fragile X ، SMA و Duchenne
مقالات مرتبط
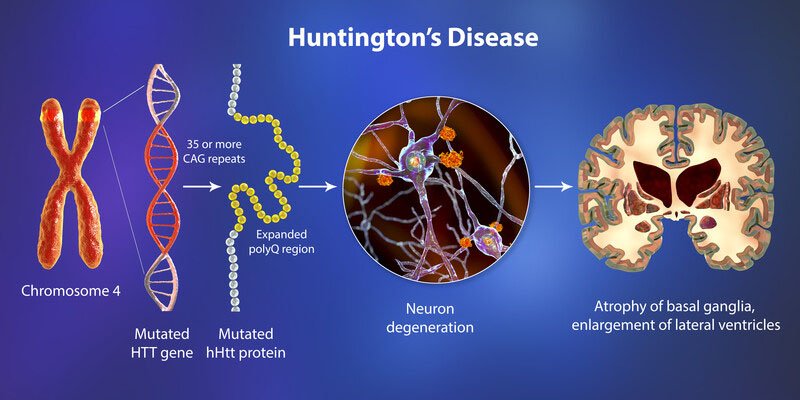
بیماری هانتینگتون و امیدی تازه برای درمان
بیماری هانتینگتون (Huntington’s Disease) یک اختلال ژنتیکی ارثی است که بهدلیل انتقال یک ژن تغییریافته از یکی از والدین به فرزند ایجاد میشود. وجود این ژنِ معیوب باعث آسیب به بخشهایی از مغز میشود و در نتیجه بر حرکت، حافظه و توانایی تفکر فرد تأثیر میگذارد.

دیستروفی عضلانی چیست ؟
دیستروفی عضلانی گروهی از بیماریهای ژنتیکی نادر است که باعث ضعف و تحلیل رفتن پیشرونده عضلات میشود. در این مقاله انواع، علائم، علل، روشهای تشخیص، درمان و راهکارهای زندگی با دیستروفی عضلانی را بررسی میکنیم.
علائم افزایش کورتیزول بدن چیست و چگونه درمان شود؟
با علائم فیزیکی و روانی افزایش کورتیزول آشنا شوید. این مقاله به بررسی نشانهها، عوارض، روشهای طبیعی کنترل کورتیزول و نقش درمان تخصصی در بهبود آن میپردازد.
بیماریهای ژنتیکی: علل، انواع و روشهای درمان
بیماریهای ژنتیکی به اختلالاتی گفته میشود که به دلیل تغییرات ژنتیکی در DNA ایجاد میشوند. در این مقاله انواع و روشهای درمان آنها بررسی میشود.

انواع اختلالات ژنتیکی در کودکان
اختلالات ژنتیکی به گروهی از بیماریها گفته میشود که در نتیجه تغییرات یا جهشهای ژنتیکی در یک یا چند ژن ایجاد میشوند.
آزمایش PGS و PGD: بارداری سالمتر
آزمایشهای Preimplantation Genetic Screening و Preimplantation Genetic Diagnosis برای زوجهایی که مشکلات ژنتیکی دارند یا به علت سن بالا یا نازایی نیاز به IVF دارند، نقش بسیار مهمی ایفا میکنند.